
- Accueil >
- Pathologies >
- Malformations ORL et cervico-faciales >
- Syndrome de Klippel-Feil
Syndrome de Klippel-Feil
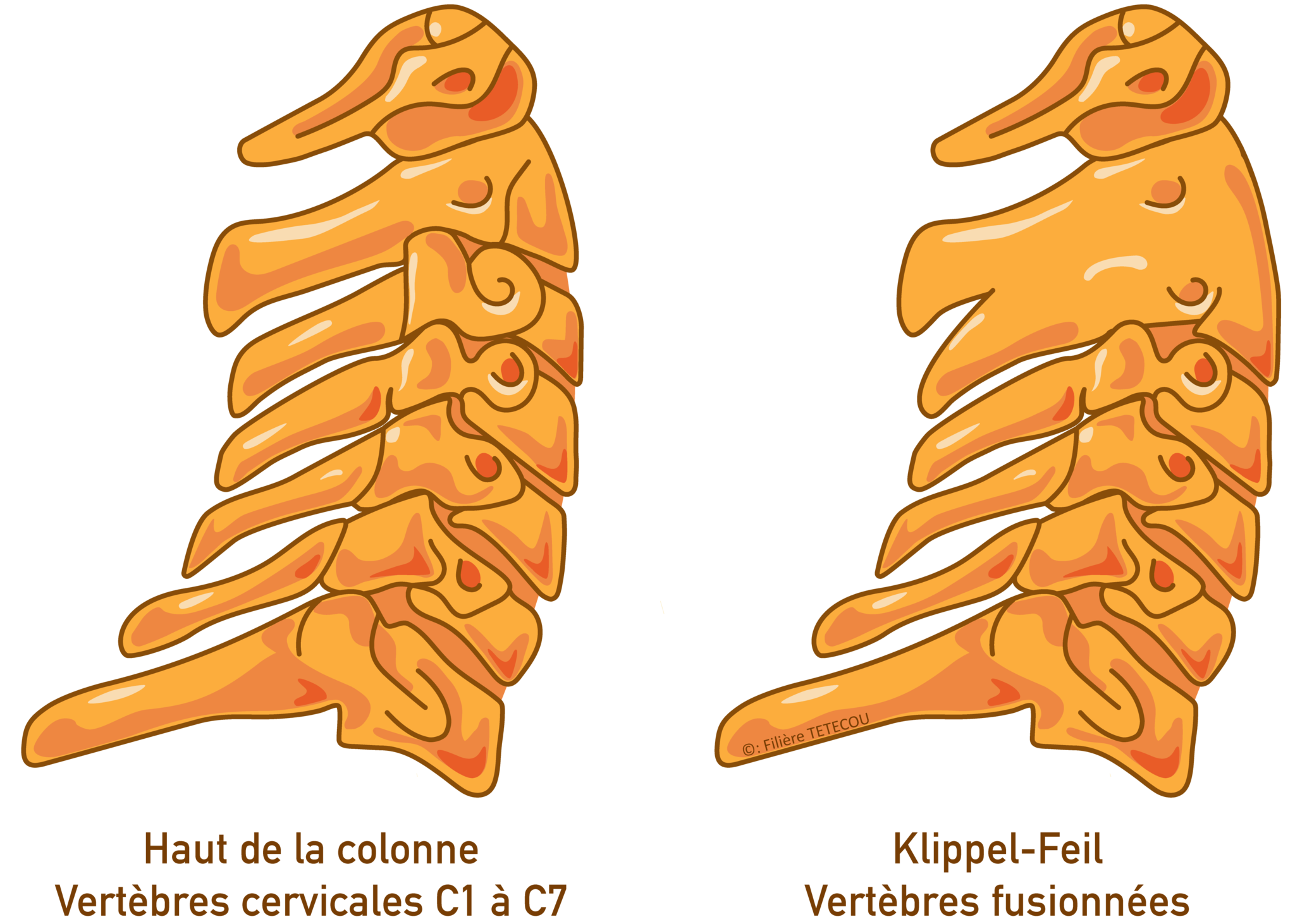
Le syndrome de Klippel-Feil est une maladie rare présente à la naissance (congénitale) caractérisée par des malformations des vertèbres du cou (fusion d'une ou plusieurs vertèbres cervicales) et se manifestant notamment par :
- un cou court,
- une mobilité restreinte de la partie supérieure de la colonne vertébrale (difficulté à pencher la tête sur le côté, vers l’avant, vers l’arrière et/ou en rotation).
Syndrome de Klippel-Feil
La prévalence du syndrome de Klippel-Feil est estimée à 1 sur 50 000.
La cause
Le syndrome de Klippel-Feil survient dans la plupart des cas de façon sporadique, c'est à dire accidentellement sans cause identifiée.
Dans de rares cas, le syndrome de Klippel-Feil peut être causé par des modifications des gènes GDF6 (localisé sur le chromosome 8 : 8q22.1) ou GDF3 (localisé sur le chromosome 12 : 12p13.31) permettant la fabrication des facteurs de croissance 3 et 6, impliqués dans le développement osseux et la segmentation des vertèbres.
D'autres gènes mutés, localisés sur les chromosomes 5 et 17 (MEOX1) seraient également impliqués.
Ces mutations génétiques sont à l'origine de la fusion d'une ou plusieurs vertèbres cervicales (synostose congénitale) lors de la formation du fœtus.
Les mutations génétiques peuvent se transmettre héréditairement, selon un mode soit :
- autosomique dominant : si l'un des parents est atteint de la maladie, c'est-à-dire qu'il a un gène normal et un gène muté, la probabilité que les fils et les filles aient la maladie est de 50 %. Les enfants qui n'ont pas reçu le gène muté n'ont pas la maladie et ne la transmettent pas.
- autosomique récessif : les deux parents sont porteurs sains d'un gène muté. À chaque grossesse, la probabilité que l'enfant reçoive le gène muté de chacun de ses parents est de 25 % ; l'enfant a alors la maladie. La probabilité que l'enfant reçoive le gène muté en un seul exemplaire est de 50 %. Alors l'enfant, comme les parents, devient un porteur sain du gène muté. La probabilité que l'enfant n'ait pas la maladie ni ne soit porteur du gène muté est de 25 %.
3 types de syndrome de Klippel-Feil ont été décrits :
- le type I impliquant plusieurs vertèbres cervicales fusionnées,
- le type II, avec la fusion d'un ou deux vertèbres, est considéré comme la forme la plus courante,
- le type III, avec des déformations de type I ou II et des déformations vertébrales thoraciques ou lombaires.
Les manifestations
Le syndrome de Klippel-Feil est à l'origine de plusieurs manifestations dont la présence et la sévérité varient d'une personne à l'autre.
-
Fusion des vertèbres et conséquences neurologiques
La fusion des vertèbres cervicales (souvent les 2e et 3e vertèbres cervicales ou C2 et C3) se manifeste par un cou court et large, une racine des cheveux basse à l'arrière de la tête, et une mobilité limitée du cou (raideur de la nuque).
Cette combinaison de manifestations caractéristiques du syndrome ne se retrouve cependant pas chez toutes les personnes concernées, certaines n'ayant qu'une mobilité réduite du cou.
Les vertèbres thoraciques et lombaires peuvent également être fusionnées.
Ces déformations vertébrales peuvent comprimer la moelle épinière d'où naissent les nerfs et alors provoquer des manifestations neurologiques tels qu'une faiblesse, un engourdissement ou des douleurs dans les bras ou les jambes.
D'autres malformations du système nerveux sont possibles, telle que :
- une malformation de Chiari de type I se caractérisant par le gonflement de la partie inférieure du cervelet à travers l'orifice de la base du crâne (appelé foramen magnum). Elle se manifeste par des maux de tête, des douleurs au cou, des étourdissements, des difficultés d'équilibre, des engourdissements dans les bras ou une faiblesse dans une moitié du corps.
- une atteinte des nerfs crâniens à l'origine d'une paralysie faciale partielle ou un effet sur les muscles oculaires.
- une moelle épinière fendue (diastématomyélie)
- une augmentation de la quantité de liquide dans le canal rachidien (syringomyélie).
-
Déformations squelettiques
Les anomalies des vertèbres peuvent être à l'origine d' :
- une inclinaison de la tête (torticolis) fréquente,
- une déviation de la colonne vertébrale (scoliose),
- omoplates hautes (déformation de Sprengel), limitant les mouvements des bras vers le haut.
Toutes ces déformations squelettiques entravent la mobilité et impactent la réalisation des actes de la vie quotidienne (s'habiller, faire sa toilette, etc.).
-
Atteintes ORL
- La perte auditive est fréquente : souvent une surdité neurosensorielle, mais parfois une surdité de transmission (anomalie de la conduction dans l'oreille moyenne).
- Une voix faible et rauque due à une paralysie des cordes vocales, causée par des lésions nerveuses.
-
Autres manifestations
De nombreuses personnes atteintes du syndrome ont une taille plus petite que celle de la population générale.
Le syndrome peut être à l'origine de traits du visage particuliers : grande distance entre les yeux (hypertélorisme), un petit menton (micrognathie), des orbites inclinées vers le bas et des oreilles basses.
Une fente palatine peut également être présente.
Près d'un tiers des personnes atteintes ont des malformation des voies urinaires ou des organes génitaux.
Des malformations cardiaques congénitales peuvent être présentes.
Le diagnostic
Le diagnostic du syndrome de Klippel-Feil repose sur l'examen clinique des manifestations : un cou court et large, une racine des cheveux postérieure basse et une mobilité restreinte du cou (triade présente chez près de la moitié des personnes atteintes) ou uniquement une raideur de la nuque.
Le diagnostic est confirmé par un examen radiographique de la colonne cervicale et de la base du crâne.
L'imagerie par résonance magnétique (IRM) de la fosse crânienne postérieure et la moelle épinière permet de visualiser toutes les malformations du cerveau ou de la moelle épinière.
Un enregistrement nocturne de la respiration et des niveaux d'oxygène dans le sang doit être effectué s'il y a des signes de troubles du sommeil.
Avec un examen échographique spécifique au cours de la grossesse, les malformations vertébrales peuvent parfois être détectées. Une analyse de mutation ciblée du placenta est possible si la mère a une mutation connue.
La prise en charge
La prise en charge globale de la personne atteinte du syndrome de Klippel-Feil et de sa famille repose sur une coopération pluridisciplinaire entre pédiatre/néonatalogue, radiologue, neurochirurgien, neurologue, orthopédiste pédiatrique, généticien, ORL, chirurgien maxillo-facial, chirurgien viscéral, gynécologue/obstétricien, urologue, cardiologue, pneumologue, anesthésiste,.. et une équipe paramédicale comprenant : kinésithérapeute, ergothérapeute, psychomotricien, audioprothésiste, psychologue, infirmier, assistant social,... en concertation avec le pédiatre ou le médecin traitant.
-
Prise en charge des anomalies squelettiques
Cette prise en charge nécessite un coopération entre neurologues et neurochirurgiens pour soulager l'instabilité cervicale ou cranio-cervicale et la compression de la moelle épinière, et pour corriger la scoliose.
Une prise en charge kinésithérapeutique permet d'améliorer la mobilité en cas d'omoplate haute (déformation de Sprengel). Si nécessaire une opération chirurgicale peut être proposée.
Il est recommandé aux personnes atteintes du syndrome de Klippel-Feil d'éviter les activités nécessitant de fléchir le cou.
-
Prise en charge neurologique
Les anomalies neurologiques (dont le Chiari) nécessitent une prise en charge par un neurologue et dans certains cas par un neurochirurgien.
-
Prise en charge ORL
Un suivi auditif précoce par un ORL est nécessaire.
La surdité peut être traitée avec des aides auditives ou une intervention chirurgicale.
Une prise en charge orthophonique est importante.
Il est possible également d'utiliser des moyens de communication alternatifs telle que la langue des signes.
-
Anesthésie
Les anesthésies, réalisées au cours des différentes interventions chirurgicales dont peuvent avoir besoin les personnes atteintes du syndrome de Klippel-Feil, nécessitent une intubation, c'est à dire l'introduction par la bouche d'un tube dans la trachée permettant de relier les poumons à une machine de ventilation
Du fait de l'immobilité et l'instabilité cervicales, des précautions particulières doivent être prises pour l'intubation, afin d'éviter des lésions neurologiques.
- Autres prises en charge
Le cœur doit être examiné par échographie et une éventuelle malformation cardiaque doit être prise en charge par un cardiologue pédiatrique.
Les malformations des voies urinaires sont également détectées par échographie et évaluées et traitées par un urologue pédiatrique.
L'Education Thérapeutique du Patient (ETP)
Les personnes atteintes du syndrome de Klippel-Feil peuvent bénéficier des programmes d'Éducation Thérapeutique du Patient suivants :
- "Éducation thérapeutique pour la prévention des complications et l'amélioration de la qualité de vie des enfants atteints de malformations de la face et de la cavité buccale" (Hôpital Necker)
- E...change de regard : Programme destiné aux enfants (entre 6 et 11 ans) ayant une maladie rare et souffrant du regard des autres (Hôpital Necker)
Des ateliers éducatifs, sur l'estime de soi par exemple, ou des rencontres avec d'autres parents ou enfants peuvent également être organisés.
Les personnes atteintes du syndrome de Klippel-Feil peuvent être prises en charge dans :
- les Centres de Référence ou de Compétence des Malformations ORL Rares (MALO) pour la prise en charge des manifestations ORL,
- les Centres de Référence ou de Compétence des Fentes et Malformations faciales (MAFACE) pour la prise en charge des malformations faciales,
- les Centres de Référence ou de Compétence des Maladies Rares Orales et Dentaires (O-Rares) pour leurs soins dentaires,


Ressources
Associations de patients
Association Française du Syndrome de Klippel-Feil
![]()
Association
Anna
![]()
Orphanet - Sites d'intérêt

Recommandations et autres références pour les professionnels





La recherche
Médias
