
- Accueil >
- Pathologies >
- Malformations faciales >
- Neurofibromatose type 1
Neurofibromatose type 1
(NF1)
La neurofibromatose de type 1 (NF1) est une maladie génétique rare, présente à la naissance (congénitale), dont les manifestations sont très variables, évolutives et sont visibles dans l’enfance et à l’adolescence.
Elle se caractérise dans la plupart des cas par la présence de taches café-au-lait sur la peau, de lentigines (taches de rousseur situées sous les bras et dans les plis de l’aine), et de tumeurs situées le long des nerfs (neurofibromes).
D’autres manifestations peuvent inclure des nodules de Lisch (petites tumeurs dans l’iris de l’œil), des tumeurs sur le nerf optique (gliome), et des déformations osseuses.
La NF1 est également appelée maladie de Von Recklinghausen.
Elle touche environ une personne sur 3000 à 4000, et autant les femmes que les hommes.
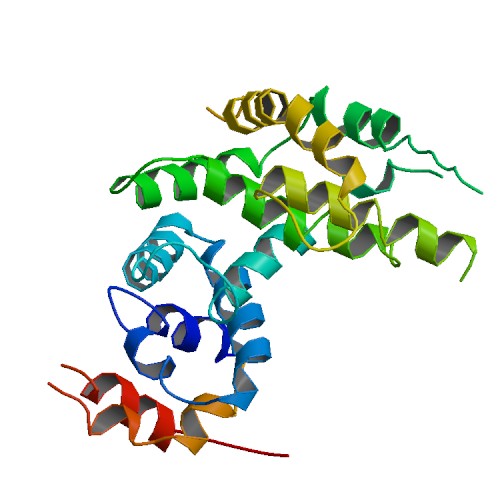
Représentation de la neurofibromine
La cause
La NF1 est due à une mutation du gène NF1, un gène suppresseur de tumeur situé sur le chromosome 17, qui code pour une protéine, la neurofibromine.
La neurofibromine intervient dans le contrôle de la multiplication des cellules, en inhibant la transmission du signal à l'intérieur des cellules.
Lors d'une mutation du gène NF1, cette protéine ne fonctionne plus ou n'est plus produite correctement, la transmission du signal n'est plus suffisamment contrôlée, ce qui est à l'origine du développement de tumeurs (neurofibromes, gliomes,...) ou d'autres anomalies (osseuses, vasculaires,...).
Dans 50 % des cas, la maladie peut être transmise par l’un des parents. C'est une transmission autosomique dominante, c'est-à-dire que le risque de transmettre la maladie à son enfant est de 1 sur 2.
Dans l'autre moitié des cas la mutation du gène NF1 apparait de façon sporadique, sans que l’un des parents ne soit atteint. On parle de néomutation ou de mutation de novo.
Transmission autosomique dominante
1 parent atteint
 Crédit : Filière AnDDi-Rares
Crédit : Filière AnDDi-Rares
Cas particulier de la NF1 mosaïque :
Il existe une forme très rare de NF1 appelée mosaïque (anciennement NF5). Dans ce cas, la mutation du gène n'est pas présente dans toutes les cellules, et les manifestations cutanées ne touchent qu'une partie du corps. Ces formes sont généralement peu sévères. Il s'agit toujours d'une néomutation.
Au sein de cette forme rare, encore plus exceptionnellement, si la mutation est aussi présente dans les cellules germinales (spermatozoides, ovocytes), la transmission de la NF1 mosaïque peut se faire d'un parent à son enfant, mais dans une proportion très inférieure à celle de 1 sur 2 comme dans la NF1 classique.
Mosaïque somatique
 Crédit : Filière AnDDi-Rares
Crédit : Filière AnDDi-Rares
Mosaïque germinale
 Crédit : Filière AnDDi-Rares
Crédit : Filière AnDDi-Rares
Les manifestations
Les manifestations possibles sont nombreuses. Elles peuvent être très légères et presque passer inaperçues, alors que d’autres porteurs de NF1 présenteront des formes sévères et des complications rares pouvant toucher plusieurs systèmes du corps humain. Cette variabilité peut se retrouver au sein d’une même famille.
Aucune personne porteuse ne présente tous les signes. Le plus souvent, seules les manifestations cutanées sont présentes. Les formes graves sont minoritaires.
La maladie est évolutive mais il n’est pas possible de prévoir son évolution. Elle peut atteindre la peau, le système nerveux, l’œil, les os et différents organes comme les poumons, le système digestif, l’appareil urinaire, les glandes endocrines et les vaisseaux. Les différentes manifestations n'apparaissent pas au même âge de la vie.
Manifestations dermatologiques
Ce sont les plus constantes et les plus précoces.
Les principales sont :
- des taches café-au-lait (TCL) : elles apparaissent dans les 3 premiers mois de vie et se multiplient jusqu’à l’âge de 2 ans. Elles s’éclaircissent chez l’adulte et peuvent même disparaitre.
- des lentigines : taches de rousseur nombreuses se situant dans les grands plis du corps (aisselles, aine, cou,…). Elles sont présentes à partir de l’âge de 4 à 6 ans.
- des neurofibromes (NF) : tumeurs bénignes (non cancéreuses) des gaines nerveuses (tissus qui entourent les nerfs) périphériques. Il en existe plusieurs types, d’évolution, de pronostic et de prise en charge différents : NF cutanés, sous-cutanés, plexiformes, internes, dysplasiques.
D’autres signes cutanés peuvent être présents (taches violines, hyperpigmentation, xanthogranulome juvénile, malformations vasculaires,…).
Autres manifestations
Certaines manifestations sont dues à la présence de neurofibromes, qui peuvent par exemple comprimer un organe, induire son mauvais fonctionnement, générer de la douleur, provoquer des atteintes neurologiques, etc.
D'autres manifestations surviennent indépendamment de la présence de ces neurofibromes.
Les manifestations non cutanées possibles comprennent :
- Des atteintes ophtalmiques : gliome des voies optiques (GVO), nodules de Lisch (petites taches pigmentées de l'iris, n'entraînant aucun symptôme masi caractéristiques de la maladie), anomalies choroïdiennes, anomalie d’une petite veinule rétinienne, ….
- Des manifestations osseuses : dysplasie (déformation) osseuse des os longs tels que le tibia, des vertèbres, du sphénoïde (os du crâne, au niveau des fosses nasales et des orbites), anomalies de la voûte crânienne, macrocéphalie, scoliose, troubles de la minéralisation osseuse,…
- Des manifestations ORL : très variables, souvent en fonction de la localisation des neurofibromes et des compressions qu'ils engendrent (neurofibromes cervicaux, laryngés, pharyngés conduisant à une apnée du sommeil, dysphagie, dyspnée), hypoacousie, atteinte de nerfs crâniens par des neurofibromes,...
- Des atteintes de la phonation : également variables en fonction de la présence ou non de neurofibromes et de leurs conséquences (dysplasies tissulaires au niveau de la langue, des gencives, altérations de la voix...)
- Des atteintes dentaires : très variables en fonction de la localisation et de l'évolution de certains neurofibromes faciaux, et des manifestations osseuses au niveau du visage
- Des manifestations endocriniennes : trouble pubertaire, retard de croissance,…
- Des manifestations cardiovasculaires : hypertension artérielle, malformations cardiaques et vasculaires
- Des manifestations pulmonaires (rares)
- Des manifestations neurologiques : trouble cognitif, le plus souvent léger, qui peut entrainer des retards d'apprentissage, céphalées, épilepsie,...
- Des cancers
- Des douleurs
- Un retentissement psychologique
Le diagnostic
Le diagnostic est principalement clinique.
Il diffère selon que le patient a également un de ses 2 parents atteint ou non.
En l’absence d'un parent atteint, le diagnostic repose sur l’association d’au moins 2 critères sur 7, qui comprennent : la présence de TCL, de lentigines, de neurofibromes, d’un gliome des voies optiques, de nodules de Lisch, de lésion osseuse et de la présence d’un variant pathogène hétérozygote du gène NF1.
Si l’un des parents est atteint, un seul de ces 7 critères est suffisant pour poser le diagnostic.
Dans certains cas, le diagnostic génétique (ou moléculaire) peut être utile, voire nécessaire, pour confirmer la maladie.
C'est pourquoi, parmi les 7 critères, celui basé sur l’analyse moléculaire du gène NF1 a été ajouté en 2021. Il permet de confirmer le diagnostic chez des patients présentant des signes inhabituels ou incomplets.
De plus, certaines manifestations, comme les TCL et lentigines, peuvent être le signe d'autres pathologies que la NF1. Dans ce cas, le diagnostic moléculaire permet de différencier de façon certaine la NF1 d’autres syndromes (syndrome de Noonan, syndrome de Legius,…), et d'établir un diagnostic plus précoce pour les patients ayant peu de manifestations dans les premières années de vie.
Des tests génétiques pourront donc être proposés au patient et à certains membres de sa famille, au cas par cas.
La prise en charge
Le traitement de la NF1 est symptomatique : il vise à traiter les manifestations et les complications de la maladie.
Il n'existe pas de traitement curatif permettant de corriger la mutation génétique qui provoque la NF1 ou de remplacer la protéine qui ne fonctionne plus.
La prise en charge est toujours pluridisciplinaire.
En fonction des manifestations, de nombreux spécialistes peuvent être impliqués et travaillent de façon coordonnée : dermatologue, chirurgien plasticien, ophtalmologue, chirurgien-dentiste, orthodontiste, ORL, orthopédiste, neurochirurgien, neurologue, pédiatre, pneumologue, cardiologue, endocrinologue, gynécologue, néphrologue, généticien, infirmier, orthophoniste, kinésithérapeute, psychomotricien, psychiatre, psychologue, médecin généraliste, cancérologue, etc.
L'évaluation initiale et le suivi doivent se faire dans un centre de référence ou de compétence labellisé pour la NF1. Le traitement repose sur une surveillance spécifique pour détecter le plus tôt possible les complications et les traiter.
En l'absence de complications, la surveillance est annuelle pour les enfants et pour les adultes ayant des risques de complications, et tous les 2-3 ans pour les adultes sans risque particulier de complications.
Les centres de la Filière TETECOU interviennent dans la prise en charge pour apporter une expertise spécifique et complémentaire, en coordination avec les centres NF1. Ainsi, les patients atteints de NF1 peuvent être pris en charge par :
-
Les chirurgiens maxillo-faciaux et plastiques des centres de référence et de compétence MAFACE pour les traitements des neurofibromes, des dysplasies du sphénoïde ou d'autres atteintes osseuses ou des tissus mous au niveau du visage,
-
Les chirurgiens ORL des centres de référence et de compétence MALO pour le traitement de certains neurofibromes touchant la sphère ORL et pour les atteintes auditives,
-
les neurochirurgiens des centres de référence et de compétence CRANIOST pour le traitement des complications de certains gliomes, la prise en charge des atteintes du crâne,
-
les chirurgiens-dentistes des centres de référence et de compétence O-Rares pour la prise en charge dentaire,
-
les pédiatres des centres de référence et de compétence SPRATON en cas de troubles de l'oralité ou de la déglutition.
![]()

![]()

![]()
Prise en charge chirurgicale des neurofibromes
-
Les neurofibromes cutanés : ces tumeurs restent bénignes et il n'y a pas de risque d'évolution cancéreuse. Elles ne sont traitées que si elles provoquent d'autres effets (douleur, saignement, démangeaisons) et/ou une gêne esthétique avec un retentissement psychologique et une diminution de la qualité de vie. Le traitement classique consiste en une éxérèse (retrait par chirurgie) et/ou une destruction par laser CO2 (en fonction du nombre, de la taille, de la localisation). D'autres traitements peuvent également être envisagés (électrochirurgie, radiofréquence,...).
-
Les neurofibromes sous-cutanés : ils sont enlevés chirurgicalement (éxérèse) seulement s'ils entrainent des conséquences sévères (douleur, altération du sens du toucher, mauvais fonctionnement d'un organe,...) ou s'ils provoquent une gêne esthétique avec des conséquences psychologiques.
-
Les neurofibromes plexiformes : leur éxérèse est réalisée précocément (dans l'enfance), avant qu'ils ne deviennent trop volumineux. L'intervention est alors plus facile, et le risque de transformation maligne est réduit. Dans certains cas, des greffes de peau (par lambeaux ou expansion cutanée), des interventions maxillo-faciales et neurochirurgicales complexes (chirurgie osseuse des mâchoires, comblement de la fosse temporale, exophtalmie) sont nécessaires.
Les éxérèses des neurofibromes sous-cutanés et plexiformes doivent être réalisées par des chirurgiens expérimentés et entraînés. La localisation de ces neurofibromes le long de trajets nerveux peut rendre les interventions complexes, notamment en raison du risque de séquelles neurologiques.
-
Les neurofibromes dysplasiques : ils sont généralement entièrement retirés, en raison du risque de transformation en tumeur maligne.
Zoom sur le traitement médicamenteux des neurofibromes plexiformes
Depuis 2021, un médicament, le sélumétinib, a obtenu une autorisation de mise sur le marché (AMM) en France et en Europe, permettant sa prescription et son remboursement chez les enfants de plus de 3 ans.
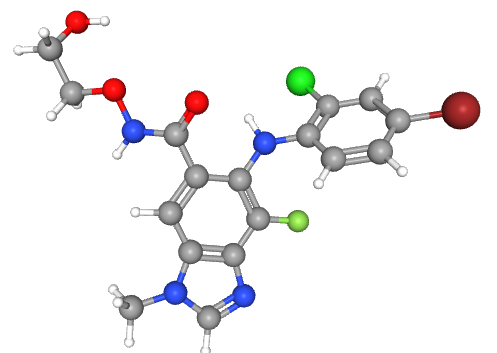
Représentation 3D du sélumétinib
(Crédit : Pubchem ID CID 10127622, https://pubchem.ncbi.nlm.nih.gov/compound/10127622)
Le sélumétinib est un anti-MEK, ou inhibiteur de MEK. Tout comme la neurofibromine, son rôle est d'inhiber la transmission du signal à l'intérieur des cellules, ce qui conduit à contrôler leur multiplication et à réduire le développement des neurofibromes.
Le sélumétinib ne remplace pas la neurofibromine car il agit à un niveau différent dans la transmission du signal, mais son action est similaire : il ralentit la prolifération cellulaire. Il remplace donc l'effet de la neurofibromine non fonctionnelle.
Il est prescrit dans des cas de neurofibromes plexiformes symptomatiques et inopérables chez l'enfant.
Il permet d'obtenir une diminution de la masse tumorale de ces neurofibromes, qui selon leur localisation en profondeur, peuvent induire des compressions d'organes. Il diminue également les douleurs qui ne peuvent pas être contrôlées par des traitements antalgiques.
En raison des effets secondaires possibles, il est cependant toujours prescrit après évaluation du bénéfice attendu et validation en réunion de concertation pluridisciplinaire (RCP) spécialisée. Une surveillance spécifique des effets secondaires et de l'efficacité sont mises en place.
Ce nouveau médicament ne remplace pas la chirurgie qui reste le traitement de référence, mais c'est une avancée majeure qui permet d'élargir l'arsenal thérapeutique et d'améliorer considérablement la vie des patients lorsque la chirurgie n'est pas possible.
Des études sont en cours pour évaluer son utilisation chez l'adulte. D'autres molécules font également l'objet d'études cliniques.
Prise en charge des principales autres manifestations
De nombreux suivis doivent être réalisés, parmi lesquels :
-
un suivi ophtalmologique rigoureux pour le dépistage du gliome des voies optiques, qui peut avoir des conséquences sur la vue,
-
un suivi des manifestations rhumatologiques et orthopédiques,
-
un suivi des retards de développement et des difficultés d'apprentissage.
En fonction des manifestations, une prise en charge adaptée est mise en place.
Prise en charge chirurgicale de la dysplasie du sphénoïde
Le traitement de la dysplasie du sphénoïde (os situé au niveau des fosses nasales et des orbites) peut s'avérer nécessaire, notamment si elle est évolutive et provoque une atteinte de l'oeil, ou si elle est liée à la présence d'u neurofibrome plexiforme qui doit être retiré. Le traitement est chirurgical et multidisciplinaire. Il implique des chirurgiens maxillo-faciaux et plastiques et des neurochirurgiens.
Prise en charge thérapeutique du gliome des voies optiques
Le GVO est une tumeur à l'intérieur du cerveau qui évolue souvent très peu et qui se transforme rarement en tumeur maligne. Elle survient essentiellement chez les enfants de moins de 6 ans. Dans la plupart des cas elle ne nécessite pas d'intervention en dehors d'une surveillance étroite.
Dans les cas minoritaires où le GVO est invasif, peut comprimer des structures et menace la vision, une prise en charge thérapeutique est mise en place. Le traitement consiste en premier lieu en une chimiothérapie, plus rarement de la chirurgie. Des traitements médicamenteux ciblés, tels que le sélumétinib, sont en cours d'étude clinique pour déterminer leur efficacité.
Cancers
La plupart des tumeurs sont bénignes, mais les patients atteints de NF1 ont un risque accru de développer un cancer.
L'ensemble des cancers est plus fréquent que dans la population générale. Les cancers des tissus mous (les tissus qui entourent et soutiennent les organes, comme la peau, la graisse, les muscles, les nerfs, les vaisseaux sanguins...) et du cerveau sont les plus fréquents.
Une surveillance à vie est requise.
Parmi les cancers des tissus mous, les tumeurs malignes des gaines nerveuses périphériques sont prises en charge en urgence dans des centres spécialisés pour les sarcomes.
Lorsqu'un GVO est présent, le risque de développer une 2e tumeur cérébrale est plus élevé. Ce risque est donc pris en compte dans la surveillance étroite des GVO.
Douleur
La douleur est très présente chez les patients avec une NF1. Elle peut commencer à un âge précoce et a de grandes conséquences sur le quotidien et la qualité de vie. Elle peut être due aux atteintes des nerfs, à la présence de neurofibromes, de déformations osseuses, du développement de tumeurs malignes, etc.
Il est très important d'évaluer régulièrement la douleur et de la prendre en charge.
La prise en charge de la douleur ne présente pas de spécificités pour la NF1 ; elle est réalisée de la même façon que pour les autres pathologies. Certaines interventions chirurgicales (éxérèse de neurofibromes,...) ou traitements médicamenteux (sélumetinib) contribuent indirectement à la prise en charge de la douleur, en diminuant ses causes.
Prise en charge psychologique, sociale et psycho-sociale
Un soutien psychologique doit être proposé aux parents dès l'annonce du diagnostic, et aux parents et patients lors de l'annonce de complications ou d'interventions lourdes.
Un conseiller en génétique peut également intervenir pour informer sur la maladie et son mode de transmission.
L'information sur les associations de patients est transmise dès les premières consultations.
Un impact psychologique et social, et une diminution de la qualité de vie, peuvent exister chez les personnes atteintes de NF1. Ils varient beaucoup d'une personne à l'autre, notamment en fonction des manifestations et des périodes de la vie : douleurs, manifestations esthétiques majeures, regard de l'autre, acceptation de la maladie, estime de soi, intégration scolaire, insertion professionnelle et sociale, nombre d'examens, d'interventions et lourdeur du suivi médical, imprévisibilité de l'évolution,...
Le dépistage, le diagnostic et la surveillance des troubles psychologiques et psychiatriques doivent être proposés tout au long de la prise en charge.
L'accompagnement social peut se faire en partenariat avec les équipes pluridisciplinaires ou avec des structures extérieures (MDPH, CPAM, services sociaux,...).
Une prise en charge en ALD hors liste peut être demandée pour les formes sévères, ou en ALD 30 en présence de tumeurs malignes. Des demandes d'allocations peuvent être réalisées, en fonction de la sévérité de l'atteinte (Allocation d'Education de l'Enfant Handicapé, Allocation Adulte Handicapé).
Education thérapeutique du patient
Les personnes atteintes de NF1 peuvent bénéficier de programmes d'Education Thérapeutique du Patient (ETP) :
-
ACTIONS-NF1 : programme d'affirmation de soi et gestion du regard de l'autre pour les enfants et adolescents affectés par une neurofibromatose de type 1 . Programme dispensé au Centre Léon Bérard à Lyon.
-
E...change de regard : programme destiné aux enfants (entre 6 et 11 ans) ayant une maladie rare et souffrant du regard des autres. Programme dispensé, entre autres, à l'Hôpital Necker, Paris.
-
Programme d'ETP NF1 du Centre de Référence CERENEF à l'Hôpital Henri Mondor, Créteil.
Ressources
Associations de patients
Centres NF1 - Orphanet - Sites d'intérêt


Documentation et livrets d'information pour les personnes malades et leurs familles



Recommandations et autres références pour les professionnels



Article Horizon NF1 - Rare à l'écoute
Prise en charge de la NF1 au site coordonnateur du CERENEF


Ressources médico-sociales



Education thérapeutique du patient




Media
Podcast "Mots de Têtes"
La neurofibromatose de type 1

Podcast "Mots de Têtes"
Un regard pour une rencontre

Podcast "FRANCHIR"
Aider à se construire au-delà du regard des autres




Recherche
