
- Accueil >
- Pathologies >
- Maladies rares orales et dentaires >
- Amélogenèse imparfaite
Amélogenèses imparfaites
Les amélogenèses imparfaites (AI) sont des maladies rares bucco-dentaires d’origine génétique, caractérisées par des dents fragiles et de couleur altérée, du fait du développement anormal de l’émail dentaire.
Ces maladies ont des conséquences non seulement fonctionnelles mais aussi esthétiques et psycho-sociales importantes.
La prévalence des amélogenèses imparfaites varie de 1/700 à 1/14 000 personnes affectées, selon les populations étudiées.
 Crédit : Cassandra Vion
Crédit : Cassandra Vion
Dentition d'un enfant atteint d'amélogenèse imparfaite hypominéralisée
La cause
L’amélogenèse désigne la formation de l’émail dentaire qui est une structure minéralisée très résistante, recouvrant les couronnes des dents sur quelques millimètres d’épaisseur conférant une solidité et une protection contre le froid/le chaud, l’acidité, les forces mécaniques de mastication.

Actuellement, plus de 100 gènes et les protéines qu’ils codent, ont été identifiés comme étant impliqués non seulement dans la formation de l’émail mais aussi dans la formation ou le fonctionnement d’autres organes.
Dans les amélogenèses imparfaites, ces gènes sont porteurs de variations qui modifient la fonction des protéines correspondantes et sont donc à l’origine d’une modification de la structure et de l’apparence de l’émail.
On distingue plusieurs formes d’AI selon le défaut qualitatif ou quantitatif de l’émail :
- AI hypoplasique (défaut quantitatif) : émail de faible épaisseur voire absent, strié et/ou avec des puits, dur, translucide, parfois rugueux ou piqueté.
 Crédit : Cassandra Vion
Crédit : Cassandra Vion
Dent atteinte d'amélogenèse imparfaite hypoplasique (coupe longitudinale)
 Crédit : Cassandra Vion
Crédit : Cassandra Vion
Dentition d'un enfant atteint d'amélogenèse imparfaite hypoplasique
- AI hypominéralisée (défaut qualitatif) : émail d’épaisseur normale, de dureté moindre et se clivant rapidement après l’éruption dentaire, de couleur jaune-brun
 Crédit : Cassandra Vion
Crédit : Cassandra Vion
Dent atteinte d'amélogenèse imparfaite hypominéralisée (coupe longitudinale)
Crédit : Cassandra Vion
Dentition d'un enfant atteint d'amélogenèse imparfaite hypominéralisée
- AI hypomature (défaut qualitatif) : émail d’épaisseur normale, relativement dur mais poreux, de couleur blanc crayeux à jaune brun
 Crédit : Cassandra Vion
Crédit : Cassandra Vion
Dent atteinte d'amélogenèse imparfaite hypomature (coupe longitudinale)
 Crédit : Cassandra Vion
Crédit : Cassandra Vion
Dentition d'un enfant atteint d'amélogenèse imparfaite hypomature
Des membres d’une même famille peuvent avoir des formes différentes d’AI.
Tout comme un même patient peut avoir plusieurs formes de défauts cliniques de l’émail (hypoplasie, hypominéralisation) sur l’ensemble de sa dentition voire sur une même dent.
L’amélogenèse imparfaite peut exister soit sous forme isolée (avec des manifestations limitées à la cavité buccale et aux dents) soit sous forme syndromique lorsqu’elle est associée à d’autres manifestations comme une épilepsie, des anomalies rénales, dermatologiques, oculaires, osseuses, etc.
Environ 120 syndromes ont été ainsi décrits dont le syndrome émail-rein, le syndrome de Jalili, le syndrome d'Heimler, le syndrome oculo-dentaire, etc.


Syndrome d'Heimler
 Crédits : Cassandra Vion (oreille et dents), Clker-Free-Vector-Images de Pixabay (oreille), FreePik (ongle)
Crédits : Cassandra Vion (oreille et dents), Clker-Free-Vector-Images de Pixabay (oreille), FreePik (ongle)
Étant donné le nombre important de gènes impliqués, ces maladies peuvent se transmettre selon différents modes (autosomique récessif, autosomique dominant, lié à X, etc.).
Des cas sporadiques (ou isolés) ont également été décrits avec un seul membre atteint au sein d’une famille.
Un conseil génétique sera proposé au patient et à sa famille par les experts des centres de référence du réseau O-Rares, en collaboration avec les médecins généticiens.
Les manifestations
Les dents temporaires ou dents de lait et les dents permanentes ou définitives peuvent être atteintes.

L’AI peut se manifester dès l’éruption de la première dent de l’enfant, à l’âge de 6 mois, avec une altération visible permettant un diagnostic et une prise en charge précoces. Elle peut ne concerner que les dents définitives.
Les personnes atteintes d’AI ont :
-
une hypersensibilité au froid, au chaud, au brossage, à la mastication, se manifestant par un inconfort ou une gêne voire des douleurs (sensibilité de la dentine) ;
-
une coloration inesthétique des dents (taches blanchâtres, dents jaunes voire brunes) générant des difficultés dans leurs relations sociales (moqueries voire harcèlement moral) qui peuvent impacter considérablement leur qualité de vie ;
-
des pertes de substance.
D’autres manifestations cliniques peuvent être présentes :
-
un risque plus important de caries ;
-
des dents de taille anormale (taurodontisme) ;
-
des dents manquantes (agénésie dentaire) ;
-
un retard d'éruption de dents ;
-
une perte prématurée de dents (résorption des racines) ;
-
une malocclusion dentaire (emboîtement anormal des dents de la mâchoire inférieure avec les dents de la mâchoire supérieure) pouvant gêner la mastication voire parfois la déglutition ainsi que l’élocution ;
-
une maladie des gencives (inflammation ou gingivite, hyperplasie) ;
-
des dents qui restent incluses.
Le diagnostic
Le diagnostic repose tout d’abord sur l’histoire familiale et médicale du patient.
L’examen clinique des anomalies de l’émail des dents temporaires et permanentes, par un chirurgien-dentiste expert, permet d'évoquer le diagnostic d’AI chez un patient et d’en caractériser le type (hypoplasique, hypominéralisé, hypomature).
Le diagnostic clinique est confirmé par un diagnostic génétique, grâce à un test génétique spécifique des anomalies bucco-dentaires (panel de gènes GenoDENT, séquençage à haut débit) proposé par un centre expert du réseau O-Rares.
Le diagnostic génétique est important pour distinguer une AI isolée d’une AI associée ou syndromique afin de mettre en place une prise en charge médicale globale des manifestations associées.
Les patients avec une forme syndromique de maladie rare à expression bucco-dentaire peuvent bénéficier d'un séquençage du génome dans le cadre d’une pré-indication du Plan France Médecine Génomique 2025.
PFMG 2025 : La pré-indication maladies rares à expressions bucco-dentaires - formes syndromiques
Pr Agnès Bloch-Zupan
Journée Nationale 2020 de la Filière TETECOU
Les amélogenèses imparfaites ne doivent pas être confondues avec certaines pathologies non rares telles que l'hypominéralisation molaires-incisives (MIH {Molar Incisor Hypomineralization} en denture permanente ou HSPM {Hypomineralized Second Primary Molars} en denture temporaire) et la fluorose.
Hypominéralisations molaires-incisives (MIH)


Fluorose


La prise en charge
La prise en charge globale du patient et de sa famille doit être la plus précoce possible et s’effectue sur plusieurs années de la petite enfance jusqu’à l’âge adulte.
Elle repose sur une coopération pluridisciplinaire entre le chirurgien-dentiste traitant, les chirurgiens-dentistes des centres spécialisés (compétents en odontologie pédiatrique, orthopédie dento-faciale, endodontie, parodontologie, réhabilitation prothétique, implantologie), le chirurgien maxillo-facial, le médecin traitant, le généticien, avec le soutien d’un psychologue.
Dans les formes syndromiques, les professionnels des spécialités médicales et paramédicales des autres organes ou systèmes affectés sont également impliqués : dermatologue, ORL, ophtalmologue, neurologue, orthophoniste, kinésithérapeute, etc…
Les patients atteints d'amélogenèses imparfaites peuvent être pris en charge dans les Centres de Référence ou de Compétence des Maladies Rares Orales et Dentaires (O-Rares) :

Le traitement
Le traitement est généralement mis en place durant l'enfance et se poursuit jusqu'à l'âge adulte.
Il a pour but de restaurer la fonction de la dentition dans la mastication, l'élocution, et de normaliser le sourire afin de favoriser le bien-être émotionnel et psychologique pour une meilleure intégration sociale, scolaire et professionnelle des patients.
Il comprend :
- la prévention avec la mise en place d’un programme de santé bucco-dentaire adapté : l’hygiène bucco-dentaire, avec un brossage correct, est très importante dans cette maladie où l’émail anormal favorise la rétention de la plaque dentaire susceptible de provoquer l’inflammation des gencives.
Un traitement préventif peut également permettre d’améliorer la minéralisation de l’émail.
- la préservation du capital dentaire, avec un suivi bucco-dentaire, au moins deux fois par an voire davantage, en fonction des manifestations de la maladie,
- les traitements restaurateurs et prothétiques pour restaurer le sourire et la fonctionnalité des dents. Selon l’âge et la situation du patient, il s’agit en particulier de :
-
coiffes pédodontiques
-
inlay-onlay
-
facettes
-
couronnes céramiques (en denture permanente)
-
résines, souvent composites, par-dessus l’émail défectueux
-
implants en remplacement des dents atteintes
-
micro-abrasion pour supprimer les défauts superficiels de l’émail
-
« éclaircissement » des dents avec des techniques d’éclaircissement en cabinet à partir de l’âge de 18 ans

- le soutien psychologique notamment pour aider les jeunes patients à gérer l’impact de cette maladie sur l’estime de soi et la confiance en soi.
L'Education Thérapeutique du Patient (ETP)
Le programme d'Éducation Thérapeutique du Patient "ETP DentO-RarEduc", destiné aux enfants et adultes atteints de maladies rares orales et dentaires, dont l'amélogenèse imparfaite, est proposé par le CRMR coordonnateur O-Rares des Hôpitaux Universitaires de Strasbourg.

Et, en lien avec ce programme d'ETP, une application destinée aux enfants présentant des anomalies dentaires et/ou orales rares sera bientôt disponible.

Ressources
Associations de personnes malades
Association
Amélogenèse France
![]()
Association
Anna
![]()
Orphanet - Sites d'intérêt

Documentation pour les personnes malades et leurs familles


Recommandations et documents pour les professionnels






La recherche
Equipe de recherche
![]()
Développements cranio-facial et bucco-dentaire, et leurs anomalies associées
Registre D[4]/Phenodent
recensement en France des patients atteints d'anomalies dentaires rares et des maladies rares associées
Quelques projets de recherche en vidéos...
Comment l'amélogenèse et la dentinogenèse imparfaites affectent-elles les adolescents sur le plan psychosocial ?
Dr Emmanuelle Noirrit-Esclassan
Journée Recherche et Innovation de la Filière TETECOU 2025
Modèles 3D récapitulant le développement dentaire pour une médecine personnalisée et la réduction de l’errance diagnostique dans les maladies rares à expressions orales
Dr Varvara Gribova
Journée Recherche et Innovation 2024 de la Filière TETECOU
Evaluation de l’intérêt de la simulation numérique pour les réhabilitations complexes de patients atteints de maladies rares orales
Dr Mélodie Clerc
Journée Recherche et Innovation de la Filière TETECOU 2022
Validation fonctionnelle de variants de signification inconnue (VUS) impliqués dans les amélogenèses imparfaites via des modèles cellulaires 3D récapitulant l’odontogenèse
Dr Isaac-Maximiliano Bugueno Valdebenito
Journée Recherche et Innovation de la Filière TETECOU 2022
L'intelligence artificielle dans le diagnostic des maladies rares à expression bucco-dentaire
Pr Agnès Bloch-Zupan
Journée Recherche et Innovation de la Filière TETECOU 2021
Quelques projets de recherche en posters...
Développement d'un modèle d'organoïde dentaire permettant la caractérisation de Variants de Signification Inconnue impliqués dans des maladies rares bucco-dentaires
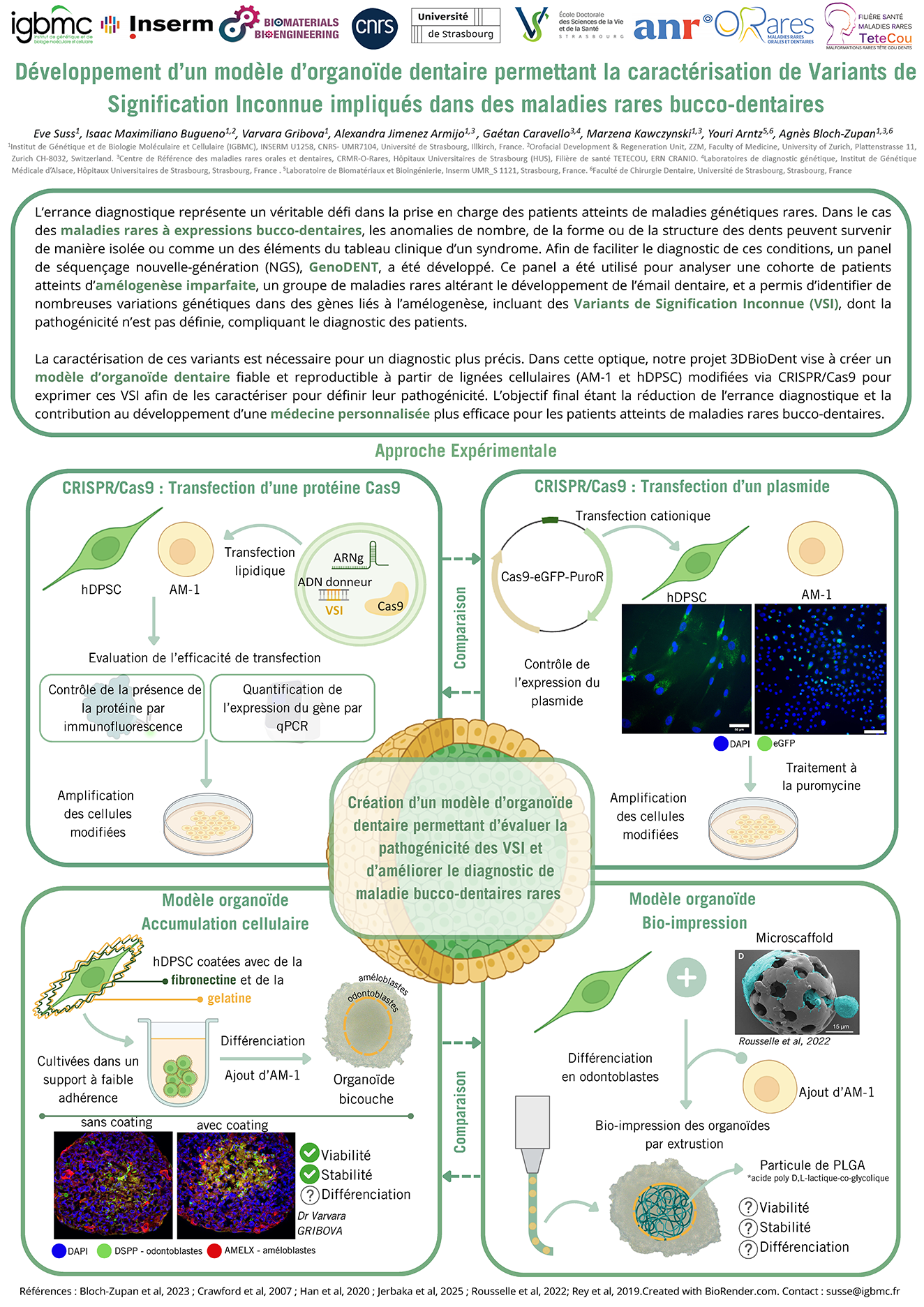
Journée Recherche et Innovation de la Filière TETECOU 2025
Implication du gène DLX3 dans les amélogenèses imparfaites isolées ou syndromiques

Journée Recherche et Innovation de la Filière TETECOU 2024
NGS leads to genotype/phenotype correlations in amelogenesis imperfecta

Journée Recherche et Innovation de la Filière TETECOU 2022
Impact psycho-social des amélo- et dentinogenèses imparfaites chez le jeune : protocole AiDiBull

Journée Recherche et Innovation de la Filière TETECOU 2021
Validation fonctionnelle de variants de signification inconnue (VUS) impliqués dans les amélogenèses imparfaites via des modèles cellulaires 3D récapitulant l’odontogenèse

Journée Nationale annuelle de la Filière TETECOU 2020
Média


Symposium O-Rares "Anomalies de l'émail"
Strasbourg, Décembre 2017
Amélogenèses imparfaites et restaurations esthétiques en céramique collée, un nouveau départ pour nos patients
Dr Olivier Etienne
Coiffes pédiatriques en zircone et anomalies de structure de l'émail du cas simple aux restaurations complexes
Dr Serena Lopez-Cazaux
Anomalies de l'émail et prévention : quelles sont les recommandations ?
Dr Magali Hernandez
Intraoral and facial 3d scanner monitoring of growth and therapy in family with amelogenesis imperfecta
Pr Tatjana Dostalova
Vidéos de l'ERN CRANIO (en anglais)
Amélogenèse imparfaite : Introduction et diagnostic
Diagnostic différentiel de l'amélogenèse imparfaite
Traitement et prise en charge de l'amélogenèse imparfaite
Comprendre la cause de l'amélogenèse imparfaite
Illustrations réalisées par Cassandra Vion, illustratrice scientifique et médicale www.cassandravion.com. Droits cédés exclusivement pour le site internet de la Filière de Santé Maladies Rares de la Tête, du Cou et des Dents (TETECOU) et de ceux de ses centres de référence (CRANIOST, MAFACE, MALO, SPRATON) qu’il héberge