
- Accueil >
- Pathologies >
- Maladies rares orales et dentaires >
- Hypophosphatasie
Hypophosphatasie
L’hypophosphatasie est une maladie génétique héréditaire rare caractérisée notamment par une fragilité osseuse et des anomalies dentaires (en particulier la perte précoce des dents).
Cette maladie est causée par des mutations du gène ALPL codant une protéine : l'isoenzyme de la phosphophatase alcaline tissulaire non-spécifique (TNSALP) ou isoenzyme ALP, dont la fonction est d'assurer la fixation du calcium, indispensable à la minéralisation des os et des dents.
Il existe six formes différentes de la maladie, selon le degré de sévérité et l'âge d'apparition avec des modes de transmission héréditaire différents.
Le nombre de personnes atteintes est estimé à 1/300 000 pour les formes sévères et 1/100 000 pour les formes modérées.
 Crédit - dent : Cassandra Vion
Crédit - os : Anatomography, CC BY-SA 2.1 JP
Crédit - scaphocéphalie : CRMR CRANIOST et Filière TETECOU
Crédit - dent : Cassandra Vion
Crédit - os : Anatomography, CC BY-SA 2.1 JP
Crédit - scaphocéphalie : CRMR CRANIOST et Filière TETECOU
Les os, le crâne et les dents sont les principales structures touchées chez les personnes atteintes d'hypophosphatasie.
Les manifestations
La maladie a des manifestations variées dont la sévérité et l'évolution sont liées au mode de transmission et au type de mutation (plusieurs centaines de mutations identifiées) déterminant l'activité de l'enzyme TNSALP résiduelle.
Le déficit ou l’absence d’activité de cette enzyme est responsable d’une anomalie de minéralisation de la plaque de croissance (partie de l’os, présente seulement à l’enfance et à l’adolescence, dont les cellules produisent la matière osseuse pour que l’os s’agrandisse), de l’os et des dents.
Le déficit de TNSALP conduit également à un déficit en vitamine B6 (pyridoxine) et une accumulation de substances responsables de manifestations neurologiques (convulsions sensibles à la pyridoxine) et musculo-articulaires (arthropathie microcristalline, fatigabilité/hypotonie musculaire).
Il existe 6 formes d'hypophosphatasie de sévérité et d'âge d'apparition différents :
- la forme périnatale (in utero) ou néonatale (à la naissance), la plus sévère, caractérisée par une insuffissance de minéralisation de tout le squelette qui peut apparaître in utero et provoquer notamment une insuffisance respiratoire à la naissance conduisant au décès du nouveau-né dans les premiers jours. Les nouveau-nés peuvent également avoir une craniosténose, c'est à dire une fermeture prématurée d’une ou plusieurs sutures crâniennes.
-
la forme périnatale bénigne, régressive, dont les manifestations squelettiques présentes in utero s’améliorent à partir du 3e trimestre et/ou pendant les premières années de vie.
-
la forme infantile (apparition avant l’âge de 6 mois), avec un déficit staturo-pondéral, un manque de tonus musculaire (hypotonie), des difficultés alimentaires, des déformations osseuses (pectus anormal, cyphoscoliose, craniosténose, genu varum), des infections broncho-pulmonaires, un rachitisme sévère à l'origine de fractures répétitives pendant la croissance, des douleurs associées.
-
la forme juvénile (apparition entre 6 mois et 18 ans), caractérisée par une chute prématurée des dents de lait (dents temporaires) avec une racine intacte, plus rarement des anomalies de la dentine ou de l’émail des dents, un retard de croissance, un rachitisme avec des fractures répétitives et inexpliquées, des troubles de la marche (retard à la marche, démarche dandinante), des douleurs musculaires et articulaires ainsi qu'une fatigabilité musculaire.
-
la forme adulte (apparition après 18 ans), caractérisée par une fragilité osseuse importante à l'origine de fractures en particulier au niveau des membres inférieurs, des atteintes articulaires, des malformations osseuses (craniosténose, incurvation des diaphyses, etc.), des douleurs diffuses, une faiblesse musculaire, une chute prématurée des dents définitives, des anomalies de l'émail dentaire, de la dentine ou des maladies parondontales touchant les tissus autour de la dent en particulier la gencive et l'os alvéolaire (parodonte).
-
la forme odonto-hypophosphatasique, sans anomalie du squelette mais uniquement des manifestations dentaires : perte précoce des dents de lait (dents temporaires) ou des dents définitives (dents permanentes), anomalies de l'émail ou de la dentine, maladies parodontales, caries dentaires, anomalies de forme ou de structure des dents temporaires ou définitives.
Les formes périnatales et infantiles sévères sont généralement transmises selon le mode récessif autosomique.
Les formes périnatale bénigne, infantile, adulte et l'odontohypophosphatasie peuvent se transmettre de manière autosomique récessive ou autosomique dominante.
Moins la forme est sévère, plus la maladie est susceptible d'avoir été transmise selon le mode autosomique dominant.
Schématiquement, plus les symptômes apparaissent tôt dans la vie, plus la maladie est sévère.
 Crédit : Cassandra Vion
Crédit : Cassandra Vion
Focus sur les anomalies dentaires
Ces anomalies touchent tous les tissus minéralisés de la dent (l’émail, la dentine, le cément et l’os alvéolaire) proportionnellement à la sévérité de la maladie.
Chez l'enfant :
La perte prématurée des dents temporaires entre 2 et 4 ans (et parfois avant), non liée à un accident (chute, ...) ou à une inflammation, avec une racine intacte est une manifestation caractéristique de la maladie. Elle est liée à une anomalie du cément (tissu permettant l’attachement de la dent à l’os alvéolaire).
La (ou les) dent(s) "bouge(nt)" (mobilité dentaire) avant de tomber (exfoliation dentaire), le plus souvent sans inflammation de la gencive ou douleurs associées.
Les incisives temporaires sont les dents les plus atteintes et le nombre et le type de dents temporaires perdues sont proportionnels à la sévérité de la maladie.
Un enfant atteint peut ainsi se retrouver progressivement partiellement, voire totalement, édenté.
Un défaut d’éruption dentaire, des anomalies de l’émail, de la dentine, de l'os alvéolaire, des polycaries et la perte précoce des dents permanentes sont également possibles chez l’enfant et l’adolescent.
Chez l'adulte :
La perte prématurée des dents peut concerner les dents permanentes, pouvant évoquer une parodontite.
Les dents permanentes peuvent présenter des défauts d’émail (stries, coloration) et de la dentine (hypominéralisation).
En cas d'odontohypophosphatasie, il existe une perte prématurée de dents temporaires ou permanentes, de nombreuses caries ou des défauts de structure de l’émail et de la dentine, une atteinte gingivale avec parodontopathie chronique.
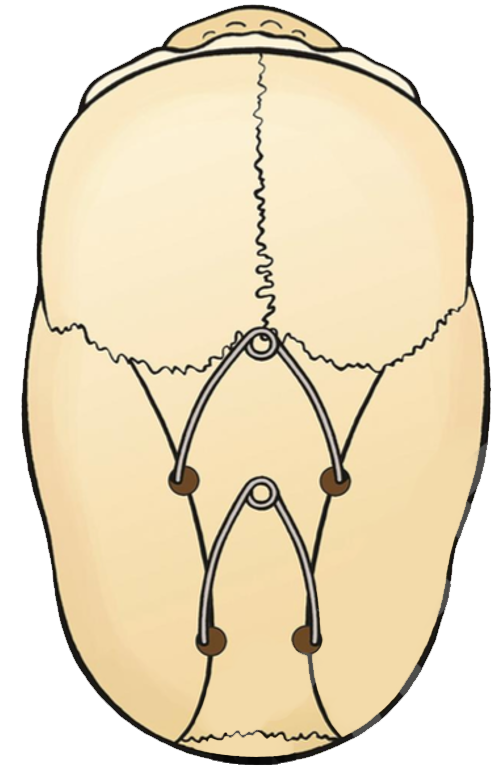
Focus sur les malformations crâniennes
L'hypophosphatasie peut être à l'origine d'une craniosténose, malformation du crâne causée par la fermeture prématurée d’une ou plusieurs sutures crâniennes.
Le plus souvent, c'est la suture sagittale qui est concernée (scaphocéphalie) ; elle peut être associée aux sutures coronales (oxycéphalie), ou à la suture lambdoïde (pansynostose).
Cette craniosténose peut provoquer une hypertension intracrânienne.
 Crédit : CRMR CRANIOST et Filière TETECOU
Crédit : CRMR CRANIOST et Filière TETECOU
Le diagnostic
Le diagnostic de l'hypophosphatasie repose sur des analyses sanguines et urinaires, des examens d’imagerie, des tests génétiques.
La maladie se traduit par une très faible activité de l’enzyme mesurable dans le sang et par la présence d’un taux de calcium élevé dans les urines.
Les manifestations bucco-dentaires sont présentes à tous les âges et stades de la maladie : les chirurgiens-dentistes peuvent être les premiers à suspecter et/ou diagnostiquer une hypophosphatasie permettant ainsi d'orienter les personnes atteintes vers les centres de référence spécialisés pour une prise en charge globale adaptée de la maladie.
La prise en charge
La prise en charge globale de la personne atteinte d'hypophosphatasie et de sa famille repose sur une coopération pluridisciplinaire entre un endocrinologue pédiatre ou un rhumatologue et/ou un médecin MPR, en concertation avec les spécialistes d’organes et le médecin traitant.
Les chirurgiens-dentistes, notamment spécialisés en odontologie pédiatrique, orthopédie dento-faciale et médecine bucco-dentaire, sont fortement impliqués dans la prise en charge.
En fonction des atteintes, les autres médecins pouvant être impliqués sont : obstétricien, néonatalogiste, médecin urgentiste et réanimateur confrontés à une complication révélatrice, médecins spécialistes confrontés à un symptôme associé (dermatologue, radiologue, neurologue, etc.), chirurgien orthopédiste, neurochirurgien, médecin de la douleur, biologiste, biochimiste, généticien clinicien et moléculaire, néphrologue et pédiatre néphrologue, chirurgien ORL, pneumologue, ophtalmologue, ...
L'équipe paramédicale comprend : infirmier (ère), conseiller en génétique, diététicien, psychologue, kinésithérapeute, orthophoniste, ergothérapeute, orthoprothésiste et prothésiste dentaire, psychomotricien, assistant social, etc.
Au sein de la Filière TETECOU, les personnes atteintes d'hypophosphatasie peuvent être prises en charge dans :
- les Centres de Référence ou de Compétence des Maladies Rares Orales et Dentaires (O-Rares) pour leurs soins dentaires,
- les Centres de Référence ou de Compétence des Craniosténoses et Malformations craniofaciales (CRANIOST) pour la prise en charge des craniosténoses
- les Centres de Référence ou de Compétence des Fentes et Malformations Faciales (MAFACE)
- les Centres de Référence ou de Compétence des Syndromes de Pierre Robin et Troubles de succion-déglutition congénitaux (SPRATON)

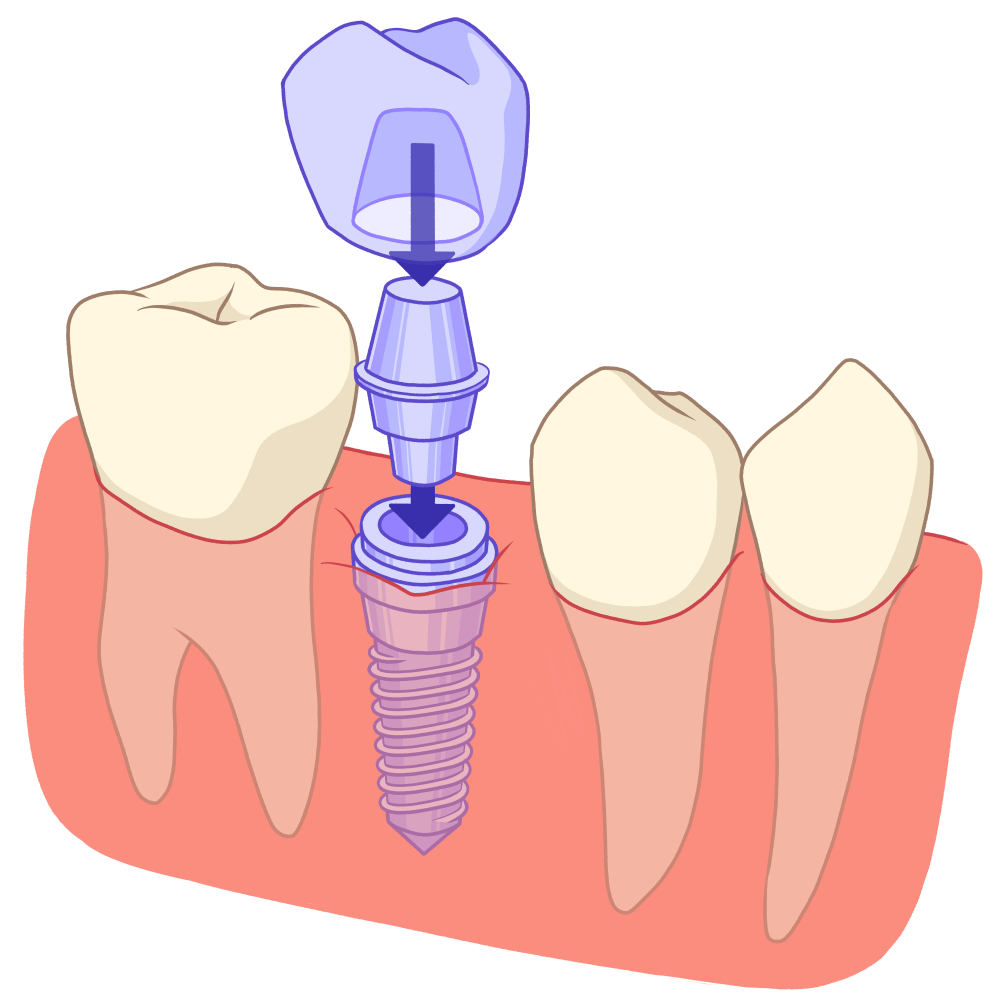 Crédit : Cassandra Vion
Crédit : Cassandra Vion
Focus sur la prise en charge dentaire
Chez l'enfant, la prise en charge bucco-dentaire a pour objectif de traiter les séquelles des pertes prématurées de dents temporaires ou permanentes et ainsi permettre une fonction masticatoire, la phonation, le sourire.
La pose de prothèses pédiatriques est réalisable dès le plus jeune âge en cas de pertes de dents.
Les anomalies de structure sont également traitées.
Chez l’adulte, une prise en charge parodontale non chirurgicale, et si besoin chirurgicale, est le plus souvent nécessaire.
Les moyens de remplacement des dents absentes, dont les prothèses sur implants dentaires, sont discutés au cas par cas et sont potentiellement réalisées précocement.
Focus sur la prise en charge craniofaciale
La craniosténose, qu'il s'agisse d'une forme dite simple (une seule suture touchée) ou complexe (plusieurs sutures concernées) nécessite une chirurgie craniofaciale, pour permettre le bon développement du cerveau.
Le traitement
Un traitement par remplacement de l'enzyme absente (enzymothérapie substitutive) peut être proposé aux personnes dont les symptômes ont commencé avant l’âge de 18 ans.
Ressources
Associations de patients
Association
Hypophosphatasie Europe
![]()
Association
Anna
![]()
Documentation et livrets d'information pour les familles
Livre
de l'Association
Hypophosphatasie Europe

Bande dessinée
de l'Association
Hypophosphatasie Europe

Livret du diagnostic
de l'Association Hypophosphatasie Europe

Orphanet et sites d'intérêt






Recommandations et autres références pour les professionnels




 janvier 2026
janvier 2026

La recherche
Registre D[4]/Phenodent
recensement en France des patients atteints d'anomalies dentaires rares et des maladies rares associées
(avec un registre spécifique dédié à l'hypophosphatasie)
Thérapie génique
Un traitement de l'hypophosphatasie par thérapie génique est en cours d'étude.
Targeting pyrophosphate dysregulation to treat soft bones and soft-tissue calcification disorders
Pr Jose Luis Millán
Journée Recherche et Innovation 2023 de la Filière TETECOU
Os, dents et médicaments orphelins
Pr Ariane Berdal
Journée internationale des maladies rares 2018
Média



Thématiques des podcasts et vidéos Rare à l'écoute
- Qu'appelle-t'on hypophosphatasie ?
- Reconnaître l'hypophosphatasie
- Diagnostiquer l'hypophosphatasie chez l'adulte
- Prendre en charge l'hypophosphatasie
- Vivre avec une hypophosphatasie
L'hypophosphatasie
Pr Martin Biosse-Duplan et Dr Maëlle Charpie
Symposium Outils Diagnostiques - OSCAR et TETECOU, le 9 mai 2023

PFMG 2025 : La pré-indication maladies rares à expressions bucco-dentaires - formes syndromiques
Pr Agnès Bloch-Zupan
Journée Nationale 2020 de la Filière TETECOU
Illustrations réalisées par Cassandra Vion, illustratrice scientifique et médicale www.cassandravion.com. Droits cédés exclusivement pour le site internet de la Filière de Santé Maladies Rares de la Tête, du Cou et des Dents (TETECOU) et de ceux de ses centres de référence (CRANIOST, MAFACE, MALO, SPRATON) qu’il héberge