
- Accueil >
- Pathologies >
- Malformations craniofaciales >
- Syndrome de Crouzon
Syndrome de Crouzon
Le syndrome de Crouzon est une maladie génétique rare présente à la naissance (congénitale) ou visible plus tard dans l'enfance. Il est caractérisée par :
- des malformations du crâne (craniosténoses : fusions prématurées de certains os du crâne au niveau de différentes sutures crâniennes)
- le développement insuffisant de la partie moyenne du visage
à l'origine :
- de malformations du visage
-
d'anomalies oculaires
- de troubles neurologiques
- de troubles ORL (auditifs, respiratoires)
- d'anomalies dentaires
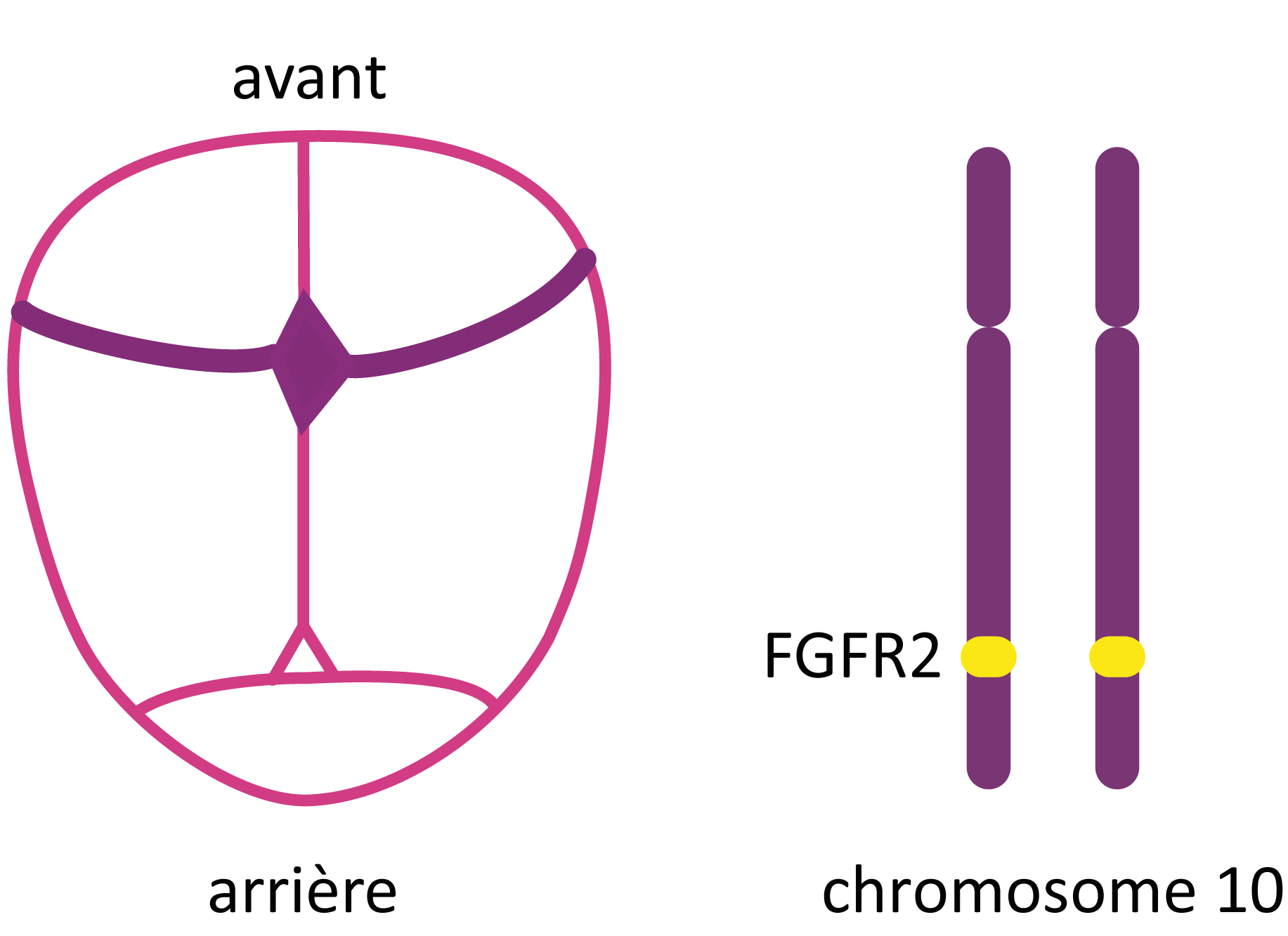
La prévalence du syndrome de Crouzon est estimée à 1 sur 50 000.
C’est une des craniosténoses syndromiques les plus fréquentes après le syndrome de Muenke et le syndrome de Saethre-Chotzen.
Le syndrome de Crouzon est dû à des mutations du gène FGFR2 localisé sur le chromosome 10 (10q26), impliqué dans la signalisation cellulaire au cours du développement embryonnaire.
Génétique des craniosténoses
Pr Corinne Collet
Journée Recherche & Innovation 2021 de la Filière TETECOU
Génétique & craniosténose
une animation de l'ERN CRANIO
Le syndrome apparaît, le plus souvent, à la suite de nouvelles mutations génétiques (néomutations) : ces cas dits sporadiques représentent 30 à 60 % des cas observés.
Le syndrome peut se transmettre héréditairement selon le mode autosomique dominant : le risque qu'un des parents atteints transmette le gène muté est de 50 %, que ce soit une fille ou un garçon.
Au sein d'une même famille, certaines personnes peuvent avoir des atteintes sévères alors que d'autres auront des manifestations plus légères.
Les manifestations
Le syndrome de Crouzon est défini comme étant une craniosténose syndromique : la craniosténose n’est pas isolée mais associée à des atteintes d’autres structures ou organes.
Il s’agit d’une faciocraniosténose, c’est-à-dire que la craniosténose est associée à des malformations du visage.
Les manifestations du syndrome peuvent être discrètes à la naissance et devenir davantage visibles au fur et mesure que l'enfant grandit.
-
Malformations du crâne
Les personnes atteintes du syndrome de Crouzon ont une craniosténose complexe (impliquant plusieurs sutures), le plus souvent bicoronale (fermeture prématurée des deux sutures coronales situées à l'avant du crâne, et qui rejoignent la fontanelle antérieure) à l'origine d'une tête large avec un front plat (on parle de brachycéphalie). D'autres sutures peuvent également être impliquées.
 Crédit : CRMR CRANIOST et Filière TETECOU
Crédit : CRMR CRANIOST et Filière TETECOU
Brachycéphalie : fermeture prématurée des deux sutures coronales
Ces malformations variables du crâne peuvent être visibles à la naissance mais le plus souvent elles apparaissent au cours de la première année de vie et évoluent jusqu'à l'âge de deux à trois ans environ.
La fusion prématurée des sutures coronales empêche la partie antérieure du crâne de se développer normalement et peut être à l'origine d'une réduction du volume intracrânien et d'une augmentation de la pression dans le crâne (hypertension intracrânienne), pouvant être responsables de conséquences importantes sur la vision et le développement psychomoteur.
Avec la croissance de l'enfant, un aspect de turricéphalie peut apparaître :
- le front est très haut et semble un peu bombé
- les orbites sont peu profondes
- l’exorbitisme devient plus évident (les yeux semblent sortir des orbites).
-
Malformations du visage
Les enfants atteints ont un développement insuffisant (hypoplasie) de la partie moyenne du visage, qui est de plus positionnée en retrait.
La mâchoire supérieure (maxillaire) est trop petite, avec une inversion de l'articulé dentaire (les dents du bas recouvrent les dents du haut).
Le front est proéminent (bosse frontale), l'os du nez est raccourci (hypoplasie) et apparaît courbé et la lèvre supérieure est fine.
La distance entre les yeux est généralement grande (hypertélorisme) et les orbites peu profondes, ce qui fait que les yeux sont saillants (exhorbitisme).
Certains enfants ont également des anomalies de la lèvre et/ou du palais (fente labiale et/ou palatine), sources de difficultés pour téter et s’alimenter.

-
Anomalies oculaires
L'hypertension intra-crânienne peut endommager le nerf optique et provoquer une perte de la vision.
Les personnes atteintes du syndrome de Crouzon ont des orbites peu profondes avec des globes oculaires qui semblent ressortir (exophtalmie).
Par conséquent, pour certains, leurs paupières ne peuvent pas se fermer, entraînant l'assèchement et l'inflammation des couches supérieures de l'œil (kératites) et également des membranes qui tapissent les surfaces internes des paupières et recouvrent le blanc des yeux (conjonctivites).
Les yeux sont plus espacés que la normale (hypertélorisme) et souvent ne pointent pas dans la même direction (strabisme).
Certaines personnes ont des mouvements oculaires rapides et involontaires (nystagmus).
-
Manifestations neurologiques
Les personnes atteintes du syndrome de Crouzon peuvent développer une hydrocéphalie, caractérisée par une altération du débit ou de l'absorption du liquide céphalo-rachidien qui circule dans les cavités du cerveau (ventricules) et du canal rachidien, et entraînant potentiellement l'augmentation de la pression des fluides dans le crâne (pression intracrânienne) et le cerveau.
Une ventriculomégalie est présente chez près de 30 % des personnes atteintes du syndrome de Crouzon.
Une anomalie de Chiari est parfois diagnostiquée.
-
Troubles ORL
Beaucoup de personnes atteintes du syndrome de Crouzon ont des malformations au niveau des voies aériennes supérieures, du pharynx et du nez.
Elles ont par conséquent des difficultés pour respirer : le ronflement est courant et certains font des pauses respiratoires durant le sommeil (apnée obstructive du sommeil de sévérité variable).
Les problèmes respiratoires peuvent survenir pendant la petite enfance et entraîner des complications potentiellement graves qui nécessitent une prise en charge ORL précoce.
Les personnes peuvent aussi avoir des difficultés pour déglutir et parler.
Les personnes atteintes du syndrome de Crouzon ont souvent un déficit auditif dû à des malformations au niveau de différentes parties de l'oreille ainsi qu'à des otites répétées.
-
Anomalies dentaires
Des anomalies dentaires sont souvent présentes : inversion de l'articulé dentaire, agénésies dentaires (le plus souvent des canines maxillaires), anomalies de l'émail, retards d'éruption, malpositions dentaires, ...
-
Autres manifestations
- malformation du squelette : fusion de vertèbres (C2 et C3 le plus souvent)
- difficultés psycho-sociales : le syndrome de Crouzon est à l'origine d'une apparence physique différente, source d'une mauvaise estime de soi avec un risque de repli sur soi, d'exclusion.
Le diagnostic
Le diagnostic peut être posé à la naissance ou pendant la petite enfance sur la base de l'examen clinique, notamment par l'observation des malformations du crâne et des traits caractéristiques du visage.
Chez certaines personnes atteintes ayant des manifestations légères, le diagnostic n'est posé qu'à l'âge adulte.
Étant donné que les malformations du crâne font également partie d'autres syndromes, des examens complémentaires peuvent être nécessaires tels qu'une radiographie du crâne pour repérer les sutures impliquées, ou un scanner ou une IRM pour déceler une hypertension intra-crânienne, une hydrocéphalie.
Les spécificités de la craniosténose permettent de différencier le syndrome de Crouzon d'autres faciocraniosténoses (syndromes de Pfeiffer, de Saethre-Chotzen, de Jackson-Weiss, de Beare-Stevenson, d'Antley-Bixler).
La confirmation du diagnostic du syndrome de Crouzon peut se faire par un test génétique moléculaire identifiant les mutations spécifiques dans le gène FGFR2.
La prise en charge
La prise en charge du syndrome de Crouzon doit être réalisée par une équipe pluridisciplinaire expérimentée de chirurgiens hautement qualifiés : neurochirurgien, chirurgien plasticien pédiatrique, chirurgien maxillo-facial, chirurgien ORL, chirurgien-dentiste ainsi que des professionnels médicaux et paramédicaux : neuropédiatre, généticien, cardiologue, ophtalmologue, pédiatre, anesthésiste, radiologue, orthodontiste, orthophoniste, nutritionniste, audioprothésiste, psychologue, puéricultrice, auxiliaire de puériculture, infirmier, assistant social.
Chez les nouveau-nés, les traitements les plus prioritaires concernent les troubles de la respiration et l'hypertension intracrânienne.
Les personnes atteintes du syndrome de Crouzon ont besoin de plusieurs interventions chirurgicales de l’enfance à l’âge adulte, qu’il est nécessaire de planifier minutieusement en collaboration avec différents spécialistes en fonction des besoins de chaque enfant, en tenant compte des manifestations et de la croissance du squelette.
Un grand nombre d'interventions chirurgicales, de l'enfance à l'âge adulte, peuvent être nécessaires, ce qui peut être souvent très éprouvant aussi bien pour la personne atteinte du syndrome que pour ses proches. Le soutien psychologique est important.
-
Prise en charge craniofaciale
Le traitement de la craniosténose est exclusivement chirurgical : il a pour but de :
- restaurer une place suffisante au cerveau en développement dans le crâne,
- diminuer une éventuelle pression intracrânienne qui peut avoir des conséquences sur le développement psychomoteur et la vision.
- améliorer l'apparence de la tête de l'enfant atteint.
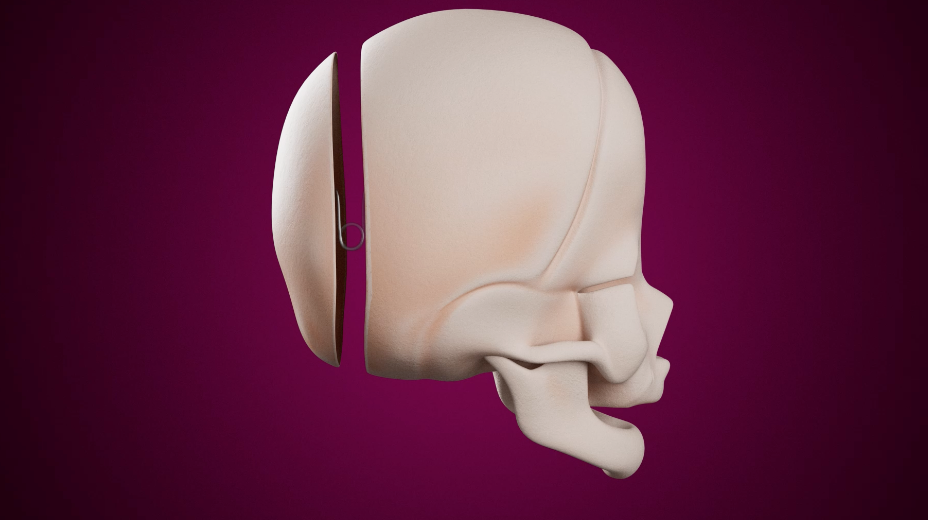
La chirurgie va permettre d’avancer la partie du front et des orbites, qui sont aplatis et déplacés vers l'arrière, avec un geste de remodelage et d'avancement fronto-crânien. La partie de front remodelée et avancée est tenue en place par des vis et des plaques résorbables qui vont disparaître 6 à 18 mois après la chirurgie.
Cette chirurgie est réalisée autour des 7-8 mois de l'enfant.
Dans certains cas, si l’enfant présente également un aplatissement postérieur du crâne, une chirurgie en 2 temps pourra être proposée :
- Un geste d’expansion postérieure avec ou sans distracteurs crâniens autour de l’âge de 5-6 mois
- Un geste d’avancement fronto-crânien vers l’âge de 9-12 mois.
La cicatrice chirurgicale est toujours la même, bien cachée dans les cheveux.
Le risque de réapparition à distance d'une hypertension intracrânienne justifie la nécessité d’un suivi annuel avec des examens ophtalmologiques de fond d'œil (FO) qui vont permettre de détecter rapidement un éventuel problème et programmer si besoin une 2e chirurgie à distance.
Un possible retard d’ossification peut justifier des chirurgies « réparatrices » à l’adolescence.
- Prise en charge ORL
Une prise en charge ORL précoce est nécessaire pour :
- traiter les troubles respiratoires
- dépister rapidement un éventuel déficit auditif en vue d'appareiller l’enfant si nécessaire, pour favoriser le développement du langage.
Elle se fait en lien avec une prise en charge orthophonique.



-
Prise en charge ophtalmologique
Une prise en charge ophtalmologique est nécessaire pour surveiller l'aspect du nerf optique et dépister un éventuel déficit visuel.
-
Prise en charge neurologique
En plus de l’IRM cérébrale, toujours réalisée au moment de la chirurgie pour bien évaluer la présence d’une éventuelle malformation du cerveau, un éléctroencéphalogramme (EEG) et une consultation avec un neuropédiatre sont nécessaires dans la première année de vie.
-
Chirurgie de la fente palatine
Les anomalies du palais sont prises en charge chirurgicalement par une équipe puridisciplinaire spécialisée (chirurgien maxillo-facial, chirurgien plasticien) avec un accompagnement orthophonique et un suivi en orthopédie dento-faciale.
-
Prise en charge dentaire
Les anomalies dentaires sont prises en charge par une équipe pluridisciplinaire spécialisée (chirurgien-dentiste, orthodontiste, etc.).
Les personnes atteintes du syndrome de Crouzon doivent être prises en charge dans les Centres de Référence ou de Compétence Maladies Rares Craniosténoses et Malformations craniofaciales (CRANIOST).
![]() Et également dans :
Et également dans :
- les Centres de Référence ou de Compétence des Fentes et Malformations faciales (MAFACE) pour la prise en charge des malformations faciales,
- les Centres de Référence ou de Compétence des Malformations ORL Rares (MALO) pour la prise en charge des manifestations ORL,
- les Centres de Référence ou de Compétence des Maladies Rares Orales et Dentaires (O-Rares) pour leurs soins dentaires,
- les Centres de Référence ou de Compétence du Syndrome de Pierre Robin et Troubles de succion-déglutition congénitaux (SPRATON) pour la prise en charge des difficultés d'alimentation.


L'Education Thérapeutique du Patient (ETP)
Les enfants porteurs du syndrome de Crouzon peuvent bénéficier des programmes suivants d'Éducation Thérapeutique du Patient au sein de la Filière TETECOU :
- Accompagnement de la distraction cranio-faciale dans les faciocraniosténoses (Hôpital Necker)
- Comprendre et mieux vivre avec une craniosténose (Hospices Civils de Lyon)
- Éducation thérapeutique pour la prévention des complications et l'amélioration de la qualité de vie des enfants atteints de malformations de la face et de la cavité buccale (Hôpital Necker)
- E...change de regard : Programme destiné aux enfants (entre 6 et 11 ans) ayant une maladie rare et souffrant du regard des autres (Hôpital Necker)
Des ateliers éducatifs, sur l'estime de soi, par exemple, ou des rencontres avec d'autres parents ou enfants peuvent également être organisés.
Ressources
Associations de personnes malades
Association Les p'tits courageux
craniosténoses syndromiques
![]()
Association Tête en l'air
enfants opérés en neurochirurgie
![]()
Association Anna
Documentation et livrets d'information pour les personnes atteintes et leurs familles


Manga "Les aventures du Lefort III"
 Crédit : Les P'tits Courageux
Crédit : Les P'tits Courageux
Education thérapeutique des enfants suivis pour une facio-craniosténose et de leur famille.
Livret d’information sur la chirurgie craniofaciale
Dr Giovanna Paternoster
Journée Nationale 2021 de la Filière TETECOU
Ressources pour les professionnels

Orphanet - Sites d'intérêt




Recommandations

PNDS
Craniosténoses syndromiques : diagnostic anténatal et prise en charge dans les six premiers mois de vie
à venir
La recherche

Les mutations activatrices dans le gène FGFR2 sont à l’origine d’anomalies de formation et de réparation osseuse
Dr Anne Morice
Journée Recherche & innovation 2023 de la Filière TETECOU
Craniofacial anomalies : lessons from animal models
Dr Laurence Legeai-Mallet
Journée Nationale 2021 de la Filière TETECOU
Modélisation de la croissance craniofaciale dans les craniosténoses - enjeux thérapeutiques
Mme Maya Geoffroy
Journée Recherche & Innovation 2021 de la Filière TETECOU
Reconnaissance des syndromes malformatifs par intelligence artificielle - premier pas vers un phénotypage multimodal
Dr Roman-Hossein Khonsari
Journée Recherche & Innovation 2021 de la Filière TETECOU
Médias




Peri-operative management of upper airway in syndromic craniosynostosis
Dr Jeremy Peuchot
Webinaire de l'ERN CRANIO, 07/11/2024
Prise en charge des patients adultes porteurs de faciocraniosténoses
Amélioration de l’annonce du diagnostic et accompagnement des patients et de leur entourage : craniosténoses
Pr Federico Di Rocco et Mme Séverine Colinet
Colloque "Recherche en Sciences humaines et sociales" de la Fondation Maladies Rares (2016)