
- Accueil >
- Pathologies >
- Malformations craniofaciales >
- Syndrome de Muenke
Syndrome de Muenke
![]() Crédit : MedlinePlus, National Library of Medicine
Crédit : MedlinePlus, National Library of Medicine
Enfant atteint d'une craniosténose coronale unilatérale
Le syndrome de Muenke associe une fusion prématurée d'une ou plusieurs sutures du crâne (le plus souvent une craniosténose bicoronale), un déficit auditif, des troubles du comportement et un risque d'épilepsie.
Des anomalies au niveau des mains et des pieds peuvent également être présentes.
La prévalence du syndrome de Muenke à la naissance est estimée à environ 1 sur 30 000 enfants, ce qui en fait la forme la plus fréquente des craniosténoses syndromiques.
Le syndrome de Muenke est une pathologie rare due à une mutation du gène FGFR3 (Pro250Arg) impliqué dans la fabrication d'une structure cellulaire nécessaire au développement normal du squelette.
Génétique des craniosténoses
Pr Corinne Collet
Journée Recherche & Innovation 2021 de la Filière TETECOU
Génétique & craniosténose
une animation de l'ERN CRANIO
Les manifestations
La plupart des personnes atteintes du syndrome de Muenke ont une craniosténose bicoronale (fermeture prématurée des deux sutures coronales) à l'origine d'une tête large avec un front plat (on parle de brachycéphalie) et trop haut (on parle de turricéphalie). Le visage est symétrique dans ces cas où les 2 sutures sont atteintes.
 Crédit : CRMR CRANIOST et Filière TETECOU
Crédit : CRMR CRANIOST et Filière TETECOU
Brachycéphalie : fermeture prématurée des deux sutures coronales
Un petit pourcentage de personnes atteintes du syndrome de Muenke ont une craniosténose coronale unilatérale. Le front est bombé d'un côté et plat de l'autre côté (on parle de plagiocéphalie) avec un visage asymétrique (asymétrie faciale) : un oeil plus petit et allongé par rapport à l'autre, une déviation de la pointe du nez, des oreilles non alignées.
 Crédit : CRMR CRANIOST et Filière TETECOU
Crédit : CRMR CRANIOST et Filière TETECOU
Plagiocéphalie : fermeture prématurée d'une seul suture coronale
 Crédit : CRMR CRANIOST, Hôpital Necker
Crédit : CRMR CRANIOST, Hôpital Necker
Enfant atteint du syndrome de Muenke
(craniosténose bicoronale)


Un déficit auditif concerne de nombreuses personnes atteintes (70%, bilatéral dans 93% des cas).
Un déficit intellectuel (40%), un risque d'épilepsie (20% ; dérèglement soudain et transitoire de l'activité électrique du cerveau se manifestant par la survenue de crises : absences, spasmes, etc.) et/ou des troubles de comportement (24%) affectent certaines personnes.
Rarement, des anomalies mineures des mains et/ou des pieds peuvent également être présentes (phalanges plus larges ou plus petites, ...).
Le diagnostic
La confirmation du diagnostic du syndrome de Muenke se fait par un test génétique moléculaire identifiant une mutation spécifique dans le gène FGFR3 (ProArg250).
La prise en charge
La prise en charge du syndrome de Muenke doit être réalisée par une équipe pluridisciplinaire expérimentée de chirurgiens hautement qualifiés : neurochirurgien, chirurgien plasticien pédiatrique, chirurgien maxillo-facial, ainsi que : neuropédiatre, généticien, ophtalmologue, ORL, pédiatre, anesthésiste, radiologue, orthodontiste, orthophoniste, psychologue, puéricultrice, auxiliaire de puériculture, infirmier, assistante sociale.
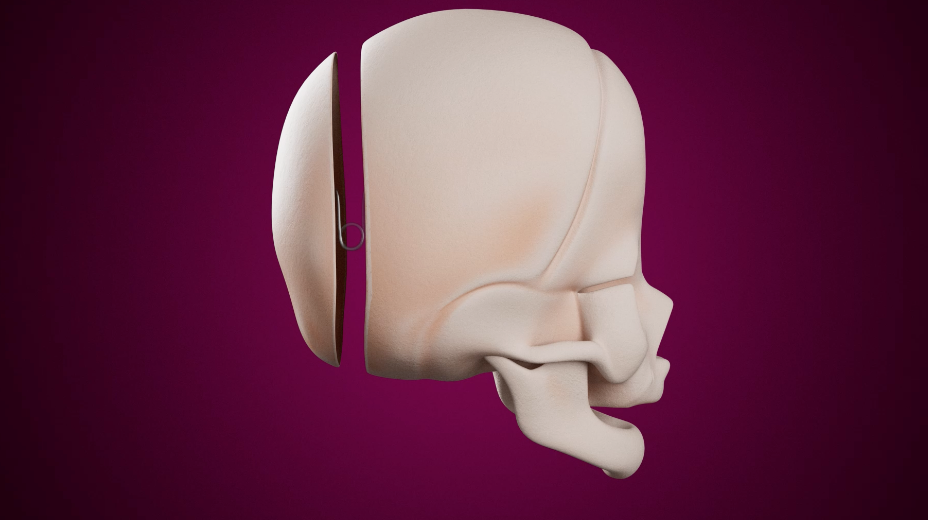
Le traitement de la craniosténose est exclusivement chirurgical et doit être réalisé dans les premiers 12 mois de vie afin d'augmenter le volume crânien et réduire ainsi le risque d’hypertension intracrânienne et les possibles conséquences sur le développement psychomoteur et la vision.
La chirurgie va permettre d’avancer la partie du front et des orbites, qui sont aplatis et déplacés postérieurement, avec un geste de remodelage et avancement fronto-crânien. La partie de front remodelée et avancée est tenue en place par des vis et des plaques résorbables qui vont disparaître 6 à 18 mois après la chirurgie.
Cette chirurgie est réalisée autour des 7-8 mois de l'enfant.
Dans certains cas, si l’enfant présente également un aplatissement postérieur du crâne, une chirurgie en 2 temps pourra être proposée :
-
Un geste d’expansion postérieure avec ou sans distracteurs crâniens autour de l’âge de 5-6 mois ;
-
Un geste d’avancement fronto-crânien vers l’âge de 9-12 mois.
La cicatrice chirurgicale est toujours la même, bien cachée dans les cheveux.
Le risque de réapparition à distance d'une hypertension intracrânienne justifie la nécessité d’un suivi annuel jusqu’à l’âge de 6 ans avec des examens ophtalmologiques de fond d'œil (FO) qui vont permettre de détecter rapidement un éventuel problème et programmer si besoin une 2e chirurgie à distance.
Un possible retard d’ossification peut justifier des chirurgies « réparatrices » à l’adolescence.
Un bilan ORL précoce est conseillé et prescrit pour pouvoir dépister rapidement l’éventuel déficit auditif et pouvoir donc appareiller l’enfant si nécessaire, pour favoriser le développement du langage.

En plus de l’IRM cérébrale, toujours réalisée au moment de la chirurgie pour bien évaluer la présence d’une éventuelle anomalie du cerveau, un éléctroencéphalogramme (EEG) et une consultation avec un neuropédiatre sont nécessaires dans la première année de vie.
"4 questions au neuropédiatre"
Docteur Marie BOURGEOIS

Neuropédiatre - Epilepsie
Neurochirurgie
Hôpital Necker-Enfants malades
- Pourquoi est-il conseillé de consulter un neuropédiatre dans le cadre du syndrome de Muenke ?
Pour s’assurer du bon développement psychomoteur des enfants atteints du syndrome, il est souhaitable d’écarter une épilepsie et un déficit auditif pour favoriser au mieux le développement et la scolarité.
Un suivi neuropédiatrique est préconisé jusqu’au collège-lycée car des aides peuvent être nécessaires pour accompagner l’enfant dans ses efforts, l’encourager, renforcer son attention, voire plus tard proposer un aménagement (tiers-temps pédagogique)
- Pourquoi faut-il faire un EEG (électroencéphalogramme, examen permettant de mesurer l'activité électrique du cerveau) à tous les enfants atteints du syndrome de Muenke ?
Car l’épilepsie peut retentir sur le développement. Un EEG de veille et sieste (électroencéphalogramme d'environ 1h comportant une période de veille et l’enregistrement d’un sommeil de sieste, couplé à un enregistrement vidéo) est préconisé comme tracé de référence si des crises devaient survenir par la suite ; il permettra de savoir si il existe des anomalies en veille et en sommeil.
- Faut-il prescrire un traitement anti-épileptique à tous les enfants atteints du syndrome de Muenke ?
Non, s'il n'y a pas de crise, un traitement anti-épileptique n'est pas nécessaire.
Les crises peuvent revêtir différentes formes : absences, crises cloniques, spasmes. Si les crises épileptiques apparaissent, il faudra refaire l’EEG.
- Comment la surdité perturbe le développement des enfants atteints du syndrome de Muenke ?
Une surdité peut être associée dans environ 1/3 des cas, il faut la dépister tôt pour éviter un retentissement sur le langage.
L'Éducation Thérapeutique du Patient (ETP)
Des programmes d'Éducation Thérapeutique du Patient peuvent être proposés aux enfants atteints du syndrome de Muenke :
- "Comprendre et mieux vivre avec une craniosténose" (Hospices Civils de Lyon)
Les personnes atteintes du syndrome de Muenke doivent être prises en charge dans les Centres de Référence ou de Compétence Maladies Rares Craniosténoses et Malformations craniofaciales (CRANIOST).
![]()
Contenu validé par le Dr Giovanna Paternoster, neurochirurgienne pédiatrique
Responsable du Centre de Référence Maladies Rares coordonnateur des craniosténoses et malformations craniofaciales CRANIOST
22 avril 2022
Ressources
Associations de personnes malades
Association Les p'tits courageux
craniosténoses syndromiques
![]()
Association Tête en l'air
enfants opérés en neurochirurgie
![]()
Association Anna
Informations pour les personnes atteintes et leurs familles


Education thérapeutique des enfants suivis pour une facio-craniosténose et de leur famille.
Livret d’information sur la chirurgie craniofaciale
Dr Giovanna Paternoster
Journée Nationale 2021 de la Filière TETECOU
Sociétés savantes - Orphanet - Sites d'intérêt




La recherche

Modélisation de la croissance craniofaciale dans les craniosténoses - enjeux thérapeutiques
Mme Maya Geoffroy
Journée Recherche & Innovation 2021 de la Filière TETECOU
Craniofacial anomalies : lessons from animal models
Dr Laurence Legeai-Mallet
Journée Nationale 2021 de la Filière TETECOU
FGFR3, un gardien de l’intégrité de l’homéostasie des sutures crâniennes : étude d’un modèle poisson zèbre perte de fonction de fgfr3
Dr Emilie DAMBROISE
Journée Recherche et Innovation 2023
Médias


Peri-operative management of upper airway in syndromic craniosynostosis
Dr Jeremy Peuchot
Webinaire de l'ERN CRANIO, 07/11/2024
Amélioration de l’annonce du diagnostic et accompagnement des patients et de leur entourage : craniosténoses
Pr Federico Di Rocco et Mme Séverine Colinet
Colloque "Recherche en Sciences humaines et sociales" de la Fondation Maladies Rares (2016)